NORMA OFICIAL MEXICANA NOM-015-SSA1-1993, Que establece las especificaciones sanitarias de los equipos para transfusión con filtro sin aguja.
Al margen un sello con el Escudo Nacional, que dice: Estados Unidos Mexicanos.- Secretaría de Salud.
AUGUSTO BONDANI GUASTI, Director General de Control de Insumos para la Salud, por acuerdo del Comité Consultivo Nacional de Normalización de Regulación y Fomento Sanitario, con fundamento en los artículos 39 de la Ley Orgánica de la Administración Pública Federal; 38, fracción II, 45, 46 fracción II, 47 de la Ley Federal sobre Metrología y Normalización; 8o. fracción IV y 12 fracción II del Reglamento Interior de la Secretaría de Salud,
INDICE
PREFACIO.
1 OBJETIVO.
2 CAMPO DE APLICACION.
3 REFERENCIAS.
4 DEFINICIONES, SIMBOLOS Y ABREVIATURAS.
5 ESPECIFICACIONES.
6 MUESTREO.
7 MARCADO Y ETIQUETADO.
8 EMPACADO.
9 METODOS DE PRUEBA.
10 BIBLIOGRAFIA.
11 OBSERVANCIA DE ESTA NORMA.
12 VIGENCIA.
PREFACIO
Las unidades administrativas que participaron en la elaboración de esta Norma son: Dirección General de Control de Insumos para la Salud; las instituciones: Instituto Mexicano del Seguro Social (Jefatura de Control de Calidad), Cámara Nacional de la Industria de la Transformación (CANACINTRA): Consejo Paramédico, Cámara Nacional de la Industria Farmaceútica (CANIFARMA) y los establecimientos siguientes: Baxter, S.A. de C.V., Abbott de México, S.A., Especialidades Científicas de Laboratorio, S.A. y Trokar, S.A. de C.V.
1. Objetivo.
Esta Norma establece las especificaciones mínimas de funcionamiento y seguridad que debe tener el equipo para transfusión con filtro, sin aguja, que se usa en los servicios médicos para la aplicación de sangre o de alguno de sus componentes, y señala los métodos de prueba para la verificación de las especificaciones.
Esta Norma tiene como finalidad reducir a un mínimo los riesgos durante la administración de sangre o de alguno de sus componentes:
- Contaminación.
- Embolismo gaseoso.
- Interacción entre el equipo de administración y los fluidos que pasan a través de él.
- Posible desprendimiento de sustancias nocivas de los materiales, de los que el equipo está construido y su paso a la circulación sanguínea.
2. Campo de aplicación.
Esta Norma Oficial Mexicana debe observarse en todas las industrias, laboratorios y establecimientos dedicados al proceso de este producto en el territorio nacional.
3. Referencias.
NOM E-097 Plásticos Acondicionamiento. Pruebas físico-químicas o de plástico, látex u otros elastómeros naturales.
NOM BB-32 Hermeticidad.
NOM BB-38 Hermeticidad.
NOM BB-8 Equipo para uso médico esterilidad. Método de Prueba.
NOM BB-6 Toxicidad, pirogenicidad y reacciones tisulares.
NOM-002-SCFI-1993 Productos envasados. Contenido neto. Tolerancias y Métodos de Verificación.
NOM Z-1 Sistema General de Unidades de Medida.
NOM 008-SCFI-1993 Sistema Internacional de Unidades (SI).
NOM Z-12/1 y Muestreo para la Inspección por
NOM Z-12/2 Atributos.
NOM Z-55 Metrología Vocabulario de términos fundamentales y generales.
2a. Edición 1992 - Ley General de Salud
1993 - Reglamento de la Ley General de Salud.
4. Definiciones, símbolos y abreviaturas.
4.1 Definiciones.
4.1.1 Anérgico: Que no reacciona con los tejidos.
4.1.2 Extremo proximal: Porción del equipo más cercano al paciente.
4.1.3 Deformación: Alteración de la forma definida.
4.1.4 Proceso: Se entiende por proceso el conjunto de actividades relativas a la obtención, elaboración, fabricación, preparación, conservación, mezclado, acondicionamiento, envasado, manipulación, transporte, distribución, importación y exportación, almacenamiento y expendio o suministro al público de los dispositivos médicos.
4.1.5 Unidad de área: Es el área seleccionada para contar las partículas; puede ser el área visual de un retículo, alguna subdivisión de ésta, una membrana cuadriculada o cualquier otra área calibrada exactamente.
4.1.6 Retículo: Conjunto de dos o más líneas cruzadas que se ponen en el foco de ciertos instrumentos ópticos y sirve para precisar lo visual o efectuar medidas muy pequeñas.
4.1.7 Hemólisis: Ruptura de los eritrocitos por efectos de agentes químicos o físicos.
4.2 Símbolos y Abreviaturas.
NOM Norma Oficial Mexicana
FEUM Farmacopea de los Estados Unidos Mexicanos
ml mililitro
mm milímetro
mm2 milímetro cuadrado
cm centímetro
cm2 centímetro cuadrado
cm3 centímetro cúbico
% por ciento
K grado Kelvin
ºC grado Celsius
kPa Kilopascal
kg/cm2 kilogramo por centímetro cuadrado
E extinción
ppm partes por millón
pH concentración de hidrogeniones
N Newton
VR Viscosidad relativa
5. Especificaciones.
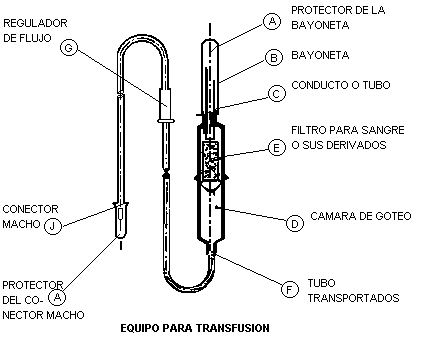
5.1 Equipo para transfusión con filtro y sin aguja. Es el dispositivo elaborado con materiales plásticos o látex flexible, atóxico, inerte y anérgico. Consta de: Protector de bayoneta, protector del conector macho, bayoneta, cámara de goteo, filtro para sangre y sus componentes, tubo transportador, regulador de flujo y conector macho (ver figura 1).
5.1.1 Protector de la bayoneta (A) y protector del conector macho (A): pieza de plástico semirrígido, las cuales deben proteger y mantener la esterilidad de la bayoneta, el conector y el interior del equipo. Deben ensamblar firmemente y removerse con facilidad (ver figura 1).
5.1.2 Bayoneta (B): pieza de plástico semirrígido, de forma cilíndrica. Uno de sus extremos debe terminar en punta. Debe tener longitudinalmente un conducto o tubo (C) el cual, comunica al recipiente que contiene el líquido a transfundir con la cámara de goteo (ver figura 1).
5.1.3 Cámara de goteo (D): pieza de plástico flexible, transparente y de forma cilíndrica ensamblada por uno de los extremos a la bayoneta y por otro a un tubo transportador. Asimismo, debe permitir un goteo continuo (ver figura 2).
5.1.4 Filtro para sangre y sus derivados (E): bolsa o tubo constituida por una malla elaborada con plástico. Uno de sus extremos debe estar cerrado. El extremo abierto debe estar ensamblado periféricamente a la pared de la cámara de goteo. El extremo cerrado no debe tener contacto con el fondo de la cámara de goteo. La malla debe tener una apertura uniforme. El diseño y ensamble del filtro puede variar, siempre y cuando no se afecte su funcionalidad.
5.1.5 Tubo transportador (F): tubo elaborado con plástico flexible y transparente. Debe ser bajo condiciones de uso, lo suficientemente flexible y resistente para adaptarse sin torceduras, colapsamientos o roturas, al equipo.
5.1.6 Regulador de flujo (G): pieza de plástico semirrígido que detenga o regule el flujo del líquido a transfundir a distintas velocidades, que una vez conseguido el ajuste de la velocidad de flujo requerida, el mecanismo permanezca firme y sin movimiento y que no dañe al tubo transportador durante el uso normal del equipo ni cuando estén almacenados en contacto.
5.1.7 Conector macho (J): pieza cónica de plástico semirrígido que debe estar ensamblada en el extremo proximal del equipo.
5.2 Especificaciones del producto.
Determinaciones.
5.2.1 Acabado: debe estar libre de fisuras, deformaciones, burbujas, oquedades, rebabas, rugosidades, roturas, desmoronamientos, material infusible, piezas faltantes, material extraño, partes chiclosas o reblandecidas, nódulos, piezas desensambladas. La cámara de goteo y tubo transportador deben ser transparentes. Todas las superficies de los componentes deben ser lisas y de color uniforme (cuando sean coloreadas).
5.2.2 Dimensiones.
5.2.2.1 Bayoneta (ver figura 3): diámetro externo de la base mayor (K) 5.5 a 6.5 mm. Diámetro externo de la base de la punta (M) 4.8 a 5.1 mm. Longitud de la base mayor a la base de la punta (N) 14 a 23 mm, a la punta (P) 24 a 29 mm.
5.2.2.2 Cámara de goteo (ver figura 3): distancia de la punta del tubo de goteo de la bayoneta al filtro (Q) 20 mm mínimo, a la salida de la cámara (R) 40 mm mínimo, a la pared de la cámara (S) 5 mm mínimo.
5.2.2.3 Filtro: área total del filtro 10 cm2 mínimo.
5.2.2.4 Tubo transportador: diámetro interno 2.7 mm mínimo. Longitud desde la cámara de goteo al conector macho 150 cm mínimo.
5.2.3 Prueba de integridad: no debe tener fuga de aire.
5.2.4 Prueba de funcionamiento.
5.2.4.1 Procedimiento A: el volumen recolectado debe ser de por lo menos 1000 ml.
5.2.4.2 Procedimiento B: el volumen recolectado debe ser de por lo menos 500 ml.
5.2.5 Partículas. Debe cumplir con la siguiente tabla.
|
Tamaño µm. |
Cantidad máxima de partículas por equipo |
|
5 a 24 |
10 |
|
25 a 49 |
5 |
|
50 a 100 |
1 |
|
Mayores de 100 |
0.5 |
|
Fibras |
0.3 |
5.2.6 Indice hemolítico: ninguna de las soluciones deben tener coloración roja. El coeficiente de extinción
1%
E1cm de la muestra problema, no debe exceder al del patrón de referencia por más de 0.03.
5.2.7 Oxido de etileno residual: 25 ppm máximo.
5.2.8 Absorbancia: La solución del extracto no debe tener una absorbancia mayor de 0.1.
5.2.9 Esterilidad: debe ser estéril.
5.2.10 Pirógenos: debe estar libre de pirógenos.
5.2.11 Prueba de seguridad: debe ser atóxico y anérgico.
5.2.12 Cambio de pH: no más de 1 ml de cualquiera de las dos soluciones volumétricas estándar, deben ser utilizados para que el indicador cambie a color gris.
5.2.13 Eficiencia del filtro: el residuo seco retenido sobre el filtro de prueba, no debe ser menor del 80% (m/m) del residuo retenido sobre el filtro de referencia.
5.2.14 Resistencia de los ensambles N (kgf): para partes pegadas 34.10 mínimo, y para partes ensambladas 14.71 mínimo.
5.2.15 Prueba al bulbo de látex: no debe tener fugas de aire (en caso de que el producto presente partes de látex deberá cumplir con las determinaciones del mismo).
5.2.16 Conicidad Luer del conector macho: debe satisfacer todas las pruebas.
5.3 Especificaciones de los plásticos.
5.3.1 Prueba intracutánea*: debe satisfacer la prueba.
5.3.2 Prueba de inyección sistémica: debe satisfacer la prueba.
5.3.3 Metales pesados: 1 ppm máximo.
5.3.4 Residuo no volátil: la diferencia entre los pesos obtenidos del extracto de la muestra y del blanco, no debe exceder de 5 mg.
5.4 Especificaciones del látex.
5.4.1 Toxicidad sistémica aguda*: debe satisfacer la prueba.
5.4.2 Prueba de reactividad intracutánea*: debe satisfacer la prueba.
5.4.3 Metales pesados: 5 ppm máximo.
5.4.4 Agentes reductores: no deben consumirse más de 2.0 ml.
6. Muestreo.
Se recomienda seguir la NOM-Z-12/1 partes 1, 2 y 3.
7. Marcado y etiquetado.
De acuerdo al artículo 210 de la Ley General de Salud.
8. Empacado.
8.1 Según la NOM-002-SCFI-1993.
9. Métodos de prueba.
9.1 Condiciones de las pruebas.
Los aparatos utilizados deben estar debidamente calibrados.
El agua empleada debe ser destilada a menos que se indique otra pureza.
El material de vidrio utilizado debe ser borosilicato de bajo coeficiente de expansión térmica.
Los reactivos utilizados deben ser grado reactivo, a menos que se indique otro grado.
* Estas pruebas no serán indispensables en producto terminado cuando existan documentos aprobatorios de las mismas en materia prima.
Utilizar un mínimo de 12 equipos para cada prueba con excepción de los métodos de prueba donde se indique el número de piezas a probar.
9.2 Acabado.
Procedimiento: inspeccionar a simple vista el producto. Debe estar libre de fisuras, deformaciones, burbujas, oquedades, rebabas, rugosidades, roturas, desmoronamientos, material infusible, piezas faltantes, material extraño, partes chiclosas o reblandecidas, nódulos, piezas desensambladas. La cámara de goteo y tubo transportador deben ser transparentes. Todas las superficies de los componentes deben ser lisas y de color uniforme (cuando sean coloreadas).
9.3 Dimensiones: utilizar un flexómetro o regla metálica para medir longitudes y un Vernier o cualquier otro instrumento adecuado, para medir diámetros.
Las muestras deben cumplir con lo establecido en el punto 5.2.2.
9.4 Prueba de integridad: sumergir el equipo para transfusión, con uno de sus extremos obturado, en un tanque de agua a una temperatura de 293 K a 303 K (20°C a 30°C). Aplicar por el extremo abierto del equipo, aire a una presión de 100 kPa (1.02 kg/cm2), arriba de la presión atmosférica durante dos minutos y observar el equipo.
No deberá tener fugas de aire.
9.5 Prueba de funcionamiento: el patrón de referencia es sangre total del mismo grupo sanguíneo, recolectada con anticoagulante, almacenada por lo menos dos semanas y que previamente se haya hecho pasar a través de un filtro con una abertura de malla de más o menos 5 mm2 o utilizar una solución de referencia, la cual debe tener la misma viscosidad relativa de la sangre (VR = 3.9 a 5.3 determinada en el viscosímetro de Hess).
9.5.1 Procedimiento A: por medio de la bayoneta, ensamblar el equipo para transfusión a un recipiente, el cual está colgado a 1 m de altura, que contenga el patrón de referencia. Abrir totalmente el regulador de flujo. Al mismo tiempo, hacer funcionar el cronómetro y determinar el volumen del patrón de referencia que fluye en 30 minutos. Recolectar el líquido en una probeta.
9.5.1.1 El volumen recolectado debe ser de por lo menos 1000 ml.
9.5.2 Procedimiento B: proceder de la misma manera que en el procedimiento A, aplicando al recipiente que contiene el patrón de referencia, una presión de 100 kPa (1.02 kg/cm2) arriba de la presión atmosférica y el tiempo de recolección es de 2 minutos.
9.5.2.1 El volumen recolectado debe ser de por lo menos 500 ml.
9.6 Determinación de partículas. Según MGA 0651 "Determinación Microscópica de Partículas" Farmacopea de los Estados Unidos Mexicanos FEUM.
9.6.1 El equipo de administración no deberá contener ninguna partícula visible cuando se observe con visión normal o lupa.
9.7 Oxido de etileno residual: consúltese la Norma NOM-BB-92 "Equipo para uso médico contenido de óxido de etileno residual" Método de Prueba.
9.8 Absorbancia:
9.8.1 Realizar esta prueba de acuerdo al método establecido en el Anexo de la Norma ISO 1135-4. La solución del extracto no debe tener una absorbancia mayor de 0.1.
9.9 Esterilidad.
Consúltese la Norma NOM-BB-8 "Equipo para Uso Médico Esterilidad". Método de Prueba.
9.10 Pirógenos.
Consúltese la Norma NOM-BB-90 "Industria Farmaceútica Determinación de pirógenos en productos médicos de un solo uso".
9.11 Prueba de seguridad: según MGA 0798 "Prueba de seguridad general para productos biológicos, Farmacopea de los Estados Unidos Mexicanos FEUM.
9.12 Prueba intracutánea según la NOM BB-42 Determinación de toxicidad, pirogenicidad y reacciones tisulares en agujas hipodérmicas desechables.
9.13 Metales pesados: consúltese la Norma NOM-BB-93 "Equipo para Uso Médico Contenido de Metales Pesados - Método Espectrofotométrico de Absorción Atómica".
9.14 Indice hemolítico.
9.14.1 Procedimiento: fundamento: se basa en la medición por espectrofotometría de la absorción de luz producida por la interacción de los grupos funcionales hemo, con energía radiante en el rango visible en función de la concentración de hemoglobina. Hemólisis es la ruptura de los eritrocitos por efecto de agentes químicos o físicos.
9.14.2 Reactivos: celdas de absorción, sangre fresca tomada de un humano en ayunas, agua inyectable, cloruro de sodio al 0.9% estéril.
9.14.3 Aparatos: centrífuga y Espectrofotómetro para registrar espectros en la región visible.
9.14.4 Preparación de reactivos, patrones y muestras.
9.14.4.1 Preparación de la suspensión de eritrocitos: diluir la sangre en una porción aproximada a cinco veces su volumen con la solución de cloruro de sodio y centrifugar por cinco minutos a una velocidad de 1500 a 2000 rpm. Decantar la solución excedente y repetir la misma operación con el paquete celular dos veces más. Contar los eritrocitos en una cámara de Neubauer o Hemocitómetro y resuspenderlos en solución de cloruro de sodio hasta obtener una concentración de 400 000 - 500 000 eritrocitos por mm3. Esta suspensión puede utilizarse por no más de 6 horas a temperatura de laboratorio.
9.14.4.2 Preparación del patrón de referencia: mezclar 5 cm3 (ml) de la solución de cloruro de sodio con 1 cm3 (ml) de la suspensión de eritrocitos preparados e incubar de 309 K a 311 K (36°C a 38°C) por 20 minutos; centrifugar durante 5 minutos a una velocidad de 1500 rpm a 2000 rpm. Utilizar el líquido sobrenadante como patrón de referencia.
9.14.4.3 Preparación de la muestra: medir bajo condiciones estériles, 20 cm3 (ml) de agua para inyección y pasarla a través de cada uno de diez equipos a probar tal y como vienen para usarse. Mezclar los efluentes y evaporarlos en baño maría.
Disolver el residuo en 5 cm3 (ml) de la solución de cloruro de sodio; adicionar 1 cm3 de la suspensión de eritrocitos preparada, mezclar y transferir a un tubo de centrífuga. Incubar de 309 K a 311 K (36°C - 38°C) por 20 minutos y centrifugar durante 5 minutos a una velocidad de 1500 a 2000 rpm. Utilizar el líquido sobrenadante como la muestra.
9.14.4.4 Procedimiento: observar las preparaciones del patrón de referencia y de la muestra y de no existir coloración roja en ninguna de las dos, ajustar el espectrofotómetro a cero de absorbancia y 100% de transmitancia, utilizando solución de cloruro de sodio como blanco. Obtener las absorbancias del patrón de referencia y de la muestra, a una longitud de onda de 576 nm o 540 nm utilizando una celda de 1 cm. Obtener los coeficientes de extinción del patrón de referencia y de la muestra por medio de la siguiente fórmula:
E1%1cm = a x 1000/bxc
donde:
a = absorbancia del patrón de referencia o de la muestra problema.
b = longitud del paso de la luz de la celda en centímetros.
c = concentración del patrón de referencia o de la muestra problema.
9.14.4.5 Interpretación: ninguna de las soluciones deben tener coloración roja. El coeficiente de extinción
1%
E1cm de la muestra problema, no debe exceder al del patrón de referencia por más de 0.03.
9.15 Toxicidad sistémica aguda: Según la NOM BB-42 "Determinación de toxicidad, pirogenicidad y reacciones tisulares en agujas hipodérmicas desechables.
9.16 Cambio de pH.
9.16.1 Preparación del extracto y blanco.
9.16.1.1 Las pruebas deben llevarse a cabo con los equipos esterilizados.
9.16.1.2 Constrúyase un sistema de circulación cerrada de tres equipos y un frasco de 300 ml de vidrio de borosilicato para ebullición. Fíjese al frasco un termostato que mantenga la temperatura del líquido en el frasco a 37°C ± 1°C. Circule 250 ml de agua para inyecciones durante 2 h por el sistema, a una velocidad de 1 L/h (usando una bomba peristáltica aplicada a un tubo adecuado de silicón, tan corto como sea posible). Colecte toda la solución y permítale enfriarse. Esta es la solución S1 (solución problema).
9.16.1.3 Utilice como blanco una alícuota de 250 ml de agua para inyecciones que se ha bombeado a través del sistema de circulación cerrado sin los equipos integrados. Esta alícuota es la solución S0 (solución Blanco).
9.16.2 Añadir 0.1 ml de la solución indicadora de Tashiro a 20 ml de la solución del extracto S1 en un recipiente para titulación.
Si el color de la solución resultante es violeta, titular con una solución volumétrica estándar de hidróxido de sodio 0.01 mol/L. Si el color de la solución resultante es gris, titular con una solución volumétrica estándar de ácido clorhídrico 0.01 mol/L hasta color gris.
Reportar el resultado en mililitros de solución de hidróxido de sodio o de solución de ácido clorhídrico utilizada.
9.16.3 La diferencia entre los mililitros utilizados en la titulación de la solución S1 y la solución S0, es el cambio de pH.
9.17 Eficiencia del filtro.
9.17.1 Equipo y reactivos: filtro de referencia con una abertura de 0.10 mm a 0.22 mm y un diámetro de hilo (monofilamento) de 0.09 mm a 0.11 mm, 4 litros de una mezcla de sangre total del mismo grupo sanguíneo con anticoagulante, almacenada por lo menos dos semanas y que previamente se haya hecho pasar a través de un filtro con una abertura de malla de más o menos 5 mm2.
9.17.2 Procedimiento: cortar dos círculos del filtro de referencia y dos círculos del filtro de prueba, los cuales deben tener cada uno, un diámetro de 40 mm. Determinar la masa (mo) de cada círculo, colocar cada círculo en un dispositivo que permita cubrir con sangre toda la superficie durante todo el periodo de prueba. Hacer pasar por gravedad un volumen de 800 ml de la mezcla de sangre a través de cada filtro. Permitir que la sangre drene completamente; posteriormente secar el filtro con el material retenido hasta aproximadamente masa constante (m1) en una estufa a temperatura de 331 K a 335 K (58°C a 62°C) y presión aproximadas de 0.65 kPa (0.0066 kg/cm2).
9.17.3 Cálculos. Por medio de la siguiente fórmula, determinar la masa del material sólido retenido en el filtro.
msr = m1 - m0
Determinar el porcentaje del material retenido en el filtro.
9.17.4 Interpretación. El residuo seco retenido sobre el filtro de prueba no debe ser menor del 80% (m/m) del residuo retenido sobre el filtro de referencia.
9.18 Resistencia de los ensambles.
9.18.1 Procedimiento: utilizar una máquina universal de pruebas mecánicas bajo las siguientes condiciones: velocidad de prueba 100 mm/mínimo, fuerza de tensión aplicada 34.10 N para partes pegadas y 14.71 N para partes ensambladas, temperaturas de 293 K a 300 K (20°C a 27°C). Tiempo empleado en la aplicación de la fuerza, 15 segundos.
9.18.2 Interpretación: las partes pegadas deben soportar la fuerza mínima de 34.10 N sin desprender o romperse. Las partes ensambladas deben soportar la fuerza mínima de 14.71 N sin desprenderse o romperse.
9.19 Prueba al bulbo de látex.
9.19.1 Procedimiento: por medio de una aguja cuyo diámetro externo sea 0.6, perforar el bulbo en el sitio indicado, sostener la aguja en posición por 15 segundos. Retirar la aguja. Sumergir el bulbo en agua y aplicar al equipo, obturando uno de sus extremos a una presión de 20 kPa arriba de la presión atmosférica por 15 segundos y observar.
9.19.2 Interpretación: no debe tener fugas de aire.
9.20 Conicidad Luer del conector macho.
9.21.1 Ver Norma NOM-BB-87/1 "Equipo para Uso Médico Conectores Cónicos con un 6% de conicidad Luer para jeringas, agujas y otros equipos médicos". Dimensiones Métodos de Prueba y la Norma NOM-BB-87/2 Parte II Conectores con sujetador roscado.
9.20.2 Interpretación: el conector cónico macho debe satisfacer todas las pruebas indicadas en la NOM-BB-87 correspondiente.
10. Bibliografía.
International Standard Transfusion Equipment for Medical Use. ISO 1135 Part 4 Transfusion Sets for Single Use.
British Standard Transfusion Equipment for Medical Use. BS 2463 Part 2, 1989 Specificatin for Administration Sets.
11. Observancia de esta Norma.
La vigilancia del cumplimiento de la presente Norma corresponde a la Secretaría de Salud, cuyo personal realizará los trabajos de verificación y vigilancia que sean necesarios.
12. Vigencia.
La presente Norma entrará en vigor con su carácter de obligatorio a partir del día siguiente de su publicación.
Sufragio Efectivo. No Reelección.
Mexico, D.F., a 21 de junio de 1994.- El Director General de Control de Insumos para la Salud, Augusto Bondani Guasti.- Rúbrica.


Fecha de publicación: 7 de noviembre de 1994